Mieux comprendre les maladies mitochondriales
D’origine génétique, une maladie mitochondriale (aussi appelée cytopathie mitochondriale) entraine un défaut de fonctionnement des mitochondries.

La mitochondrie, centrale énergétique des cellules
Chaque cellule de l’organisme – à l’exception du globule rouge – comporte de nombreuses mitochondries pour fournir l’énergie nécessaire à son bon fonctionnement. Grâce à l’oxygène respiré et au prix de nombreuses réactions chimiques, ces petits organites transforment les nutriments issus de l’alimentation en ATP (adénosine-tri-phosphate) qui est le « carburant » cellulaire.
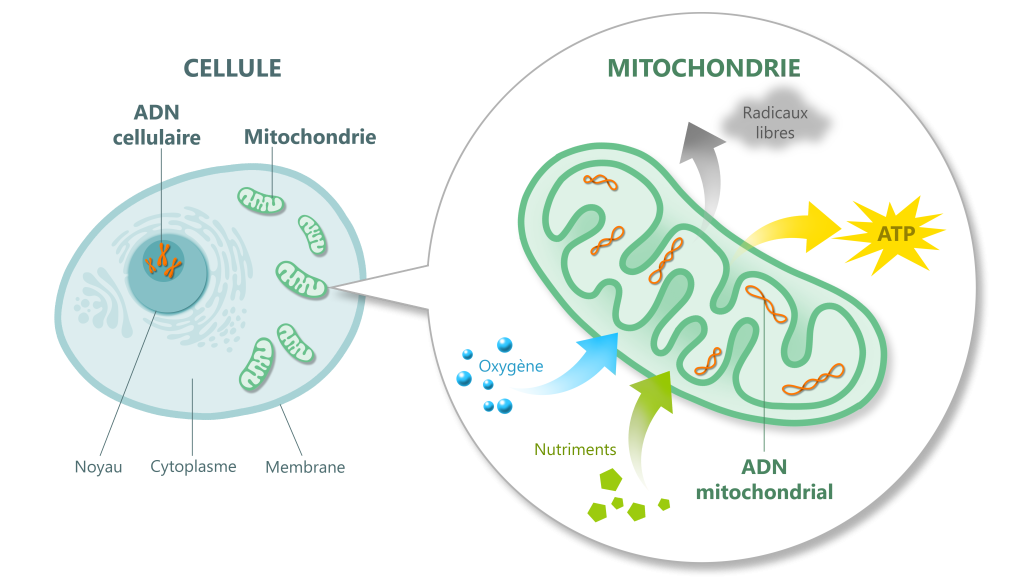
Plus un tissu a besoin d’énergie, plus le nombre de mitochondries est élevé, variant de quelques centaines à quelques milliers par cellule. Une cellule cardiaque ou musculaire squelettique compte ainsi plusieurs milliers de mitochondries.
Revers de la médaille, cette production d’énergie s’accompagne de celle de radicaux libres. Ces molécules oxydantes agissent comme la fumée d’une centrale énergétique : en trop grande quantité, ces composés peuvent être « toxiques » pour l’ensemble des constituants de la cellule.
Enfin, la mitochondrie est le seul organite à comporter son propre ADN (ADNmt), en plusieurs copies (de 2 à 10) par mitochondrie. Celui-ci est composé de 37 gènes qui codent des protéines nécessaires au fonctionnement de la mitochondrie. Cependant, la très grande majorité des protéines mitochondriales (environ 1 500) est codée par l’ADN nucléaire de la cellule.
Une grande famille
Les maladies mitochondriales affectent le fonctionnement de la très grande majorité de nos organes et tissus. Elles peuvent se manifester à toutes les périodes de la vie : à la naissance, pendant la petite enfance, l’adolescence ou à l’âge adulte. Elles touchent environ une personne sur 4 300 et sont dues à des anomalies soit de l’ADN mitochondrial (15 à 20% des cas), soit de l’ADN nucléaire.
Cette grande diversité des symptômes mais aussi cette double origine génétique complique le diagnostic, le conseil génétique et le développement de traitements.
Le saviez-vous ?
Une mutation donnée de l’ADN mitochondrial n’est en général présente que dans certaines mitochondries d’une même cellule, et non dans toutes les mitochondries. Cette coexistence d’organites mutés et non mutés au sein d’une cellule est appelée « hétéroplasmie » mitochondriale. Elle influence le risque de transmission de l’anomalie génétique à ses enfants, mais aussi la gravité de la maladie qui dépend de la proportion de molécules d’ADN mitochondriales mutées. Le taux d’hétéroplasmie peut varier d’un tissu à l’autre, ce qui peut rendre difficile à détecter l’anomalie génétique et ainsi retarder le diagnostic. Il est donc habituel de déterminer la présence ou pas d’un variant génétique, non pas dans un seul tissu mais dans plusieurs, en particulier à partir de l’ADN extrait des cellules urinaires du malade, qui représente le tissu électif (après le tissu musculaire) pour la détection ou la confirmation de la présence d’un variant de l’ADN mitochondrial.

Les mitochondries étant présentes dans presque toutes les cellules, une même anomalie génétique peut impacter un ou plusieurs tissus et entraîner des symptômes très différents, certains étant néanmoins plus marqués que d’autres selon les pathologies. Les organes susceptibles d’être touchés sont essentiellement les tissus fortement consommateurs d’énergie tels que muscles (faiblesse musculaire), le cerveau (troubles de l’apprentissage, de l’attention, épilepsie…), le cœur (cardiomyopathie, troubles de la conduction cardiaque), les yeux (atteinte de la rétine, atrophie du nerf optique), les oreilles (surdité), les reins et l’appareil digestif (difficultés à avaler, diarrhée ou constipation…).
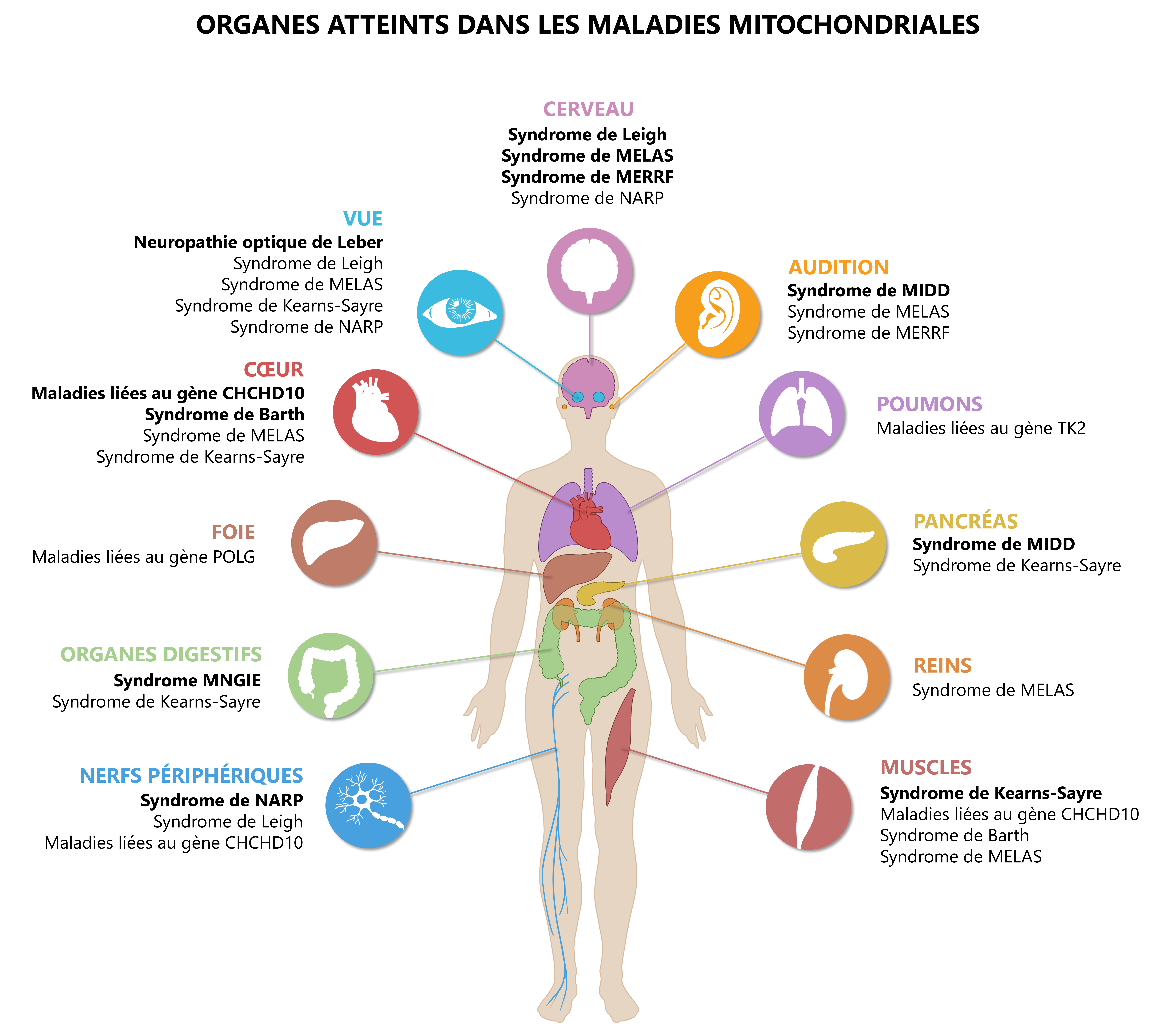

La gravité des symptômes est variable d’une maladie mitochondriale à l’autre, et parfois d’une personne à l’autre pour une même maladie. L’évolution est le plus souvent lente, avec toutefois de possibles phases d’exacerbation des manifestations liées à l’importance des efforts produits, à une infection, une opération…
Penser à une maladie mitochondriale, un premier pas vers le diagnostic
Symptômes variés, inexpliqués, sans rapport évident les uns avec les autres… Dans ce type de situations, il est important de s’interroger : et si c’était une maladie mitochondriale ? Des médecins experts pourront répondre.
Trouver un centre spécialisé
Un risque de transmission génétique très variable
Les chercheurs ont déjà identifié à ce jour plus de 400 gènes responsables de maladies mitochondriales. Ils peuvent se localiser :
- sur les chromosomes du noyau de la cellule (ADN nucléaire), le gène muté peut alors se transmettre à la descendance selon différents modes de transmission (autosomique dominant, récessif, lié à l’X), en fonction de la maladie mitochondriale,
- sur l’ADN mitochondrial, comme dans les syndromes de MELAS et de MERRF. La mutation génétique ne peut alors être transmise que par la mère car seules les mitochondries (et leur ADN) de la maman sont transmises durant l’étape de fécondation, pas celles du papa.
À retenir
Un homme n’est pas transmetteur à sa descendance d’une mutation de l’ADN mitochondrial. Seule une femme pourra éventuellement la transmettre.
Une consultation pour un conseil génétique aide à préciser le risque de transmission de la mutation génétique aux futurs enfants d’une personne qui est porteuse.